
Fold proteins and biomolecular complexes with ESMFold2
ESMFold2 is a state-of-the-art model for biomolecular complex structure prediction, developed by Biohub and released under an open license. Built on ESMC representations, it produces leading accuracy for protein-protein and antibody-antigen interactions at any given compute budget.
ESMFold2 is available in two configurations:
- ESMFold2: the larger model for maximum accuracy. It can be run either from a single sequence or with MSA context, with MSAs improving performance on difficult complexes.
- ESMFold2-Fast: a smaller model optimized for very fast single-sequence folding. It is well suited for high-throughput folding, designed sequences, metagenomic proteins, and targets with limited homologous sequence information.
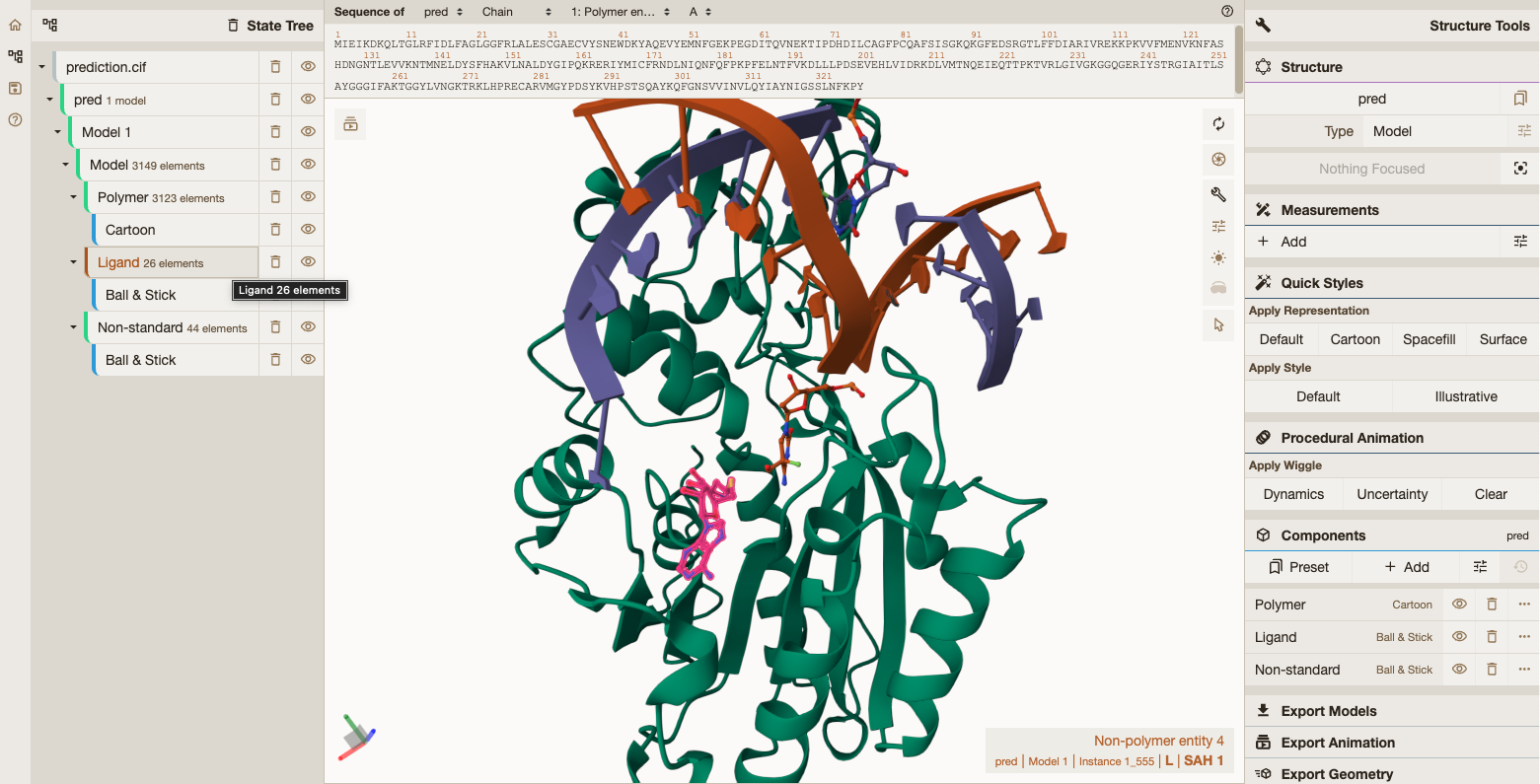
In this example, we demonstrate how to run ESMFold2 on Modal’s flexible serverless infrastructure. By default, we fold a protein-DNA-ligand complex (the M.HhaI DNA methyltransferase bound to a methylated DNA duplex and its SAH cofactor), which exercises the model’s full multimer capabilities. You can also pass any single-chain protein sequence from the command line.
This script is meant as a starting point that demonstrates how to
create a modal.Image with the correct dependencies, cache weights to a modal.Volume,
and save the output to a file for a single folding request.
To really leverage Modal’s serverless infrastructure, try scaling inference up across
hundreds or thousands of structures or invert the model to design binders
for a target protein.
Setup
from pathlib import Path
from typing import Optional
import modal
here = Path(__file__).parent # the directory of this file
MINUTES = 60 # seconds
app = modal.App(name="example-esmfold2")Installing ESMFold2 Python dependencies on Modal
Code executing on Modal runs inside containers built from modal.Images that include that
code’s dependencies.
For ESMFold2, we only need the esm library from Biohub which will install the necessary dependencies
including a custom fork of the transformers library.
ESM_REVISION = "81b3646c9429ea8458918415ad6a46178cb59833" # pin upstream commit so builds are reproducible
esmfold2_image = (
modal.Image.debian_slim(python_version="3.13")
.apt_install("git")
.uv_pip_install(
f"esm @ git+https://github.com/Biohub/esm.git@{ESM_REVISION}",
)
)We’ll use the image.imports() context manager to import libraries we’ll need in our inference code.
The context manager allows us to import libraries that might not be installed locally but are installed in our modal.Image.
with esmfold2_image.imports():
from esm.models.esmfold2 import (
DNAInput,
ESMFold2InputBuilder,
LigandInput,
Modification,
ProteinInput,
StructurePredictionInput,
)
from transformers.models.esmfold2.modeling_esmfold2 import ESMFold2ModelCaching ESMFold2 model weights on Modal Volumes
Rather than re-downloading the model weights on each cold start, we cache them on a Modal Volume. The first time you run inference, you’ll see that downloading the weights takes several minutes, but subsequent runs will start up significantly faster. For more on storing model weights on Modal, see this guide.
esmfold2_volume = modal.Volume.from_name("esmfold2-models", create_if_missing=True)
models_dir = Path("/models")We also need to point the HF cache at the Volume, and we’ll enable high-performance downloads by setting some environment variables on our modal.Image.
esmfold2_image = esmfold2_image.env(
{
"HF_HOME": str(models_dir),
"HF_XET_HIGH_PERFORMANCE": "1", # speed up downloads
}
)Running ESMFold2 on Modal
To run inference on Modal, we define an ESMFold2Inference class and wrap it with the @app.cls decorator.
The decorator takes some arguments that describe the infrastructure
our code needs to run: the Volume we created, the Image we defined, and of
course a GPU. We’ll use an H100, but you can use any other GPU supported by Modal.
When we use the @app.cls decorator, we can define a method decorated with the @modal.enter() lifecycle hook.
This method will be run once when a new container starts.
The exeuction time of the @modal.enter() method is included in the container startup time, so it won’t serve requests
until it’s ready.
To enable remote execution, we decorate our fold method with @modal.method(). We’ll demonstrate later how to call it using fold.remote().
ESMFOLD2_REPO = "biohub/ESMFold2"
ESMFOLD2_REVISION = "6234905" # pin for reproducibility
@app.cls(
image=esmfold2_image,
volumes={models_dir: esmfold2_volume},
gpu="H100",
timeout=20 * MINUTES,
)
class ESMFold2Inference:
@modal.enter()
def load_model(self):
print("🧬 loading ESMFold2 onto the GPU")
self.model = (
ESMFold2Model.from_pretrained(ESMFOLD2_REPO, revision=ESMFOLD2_REVISION)
.cuda()
.eval()
)
@modal.method()
def fold(
self,
sequence: Optional[str] = None,
num_loops: int = 3,
num_sampling_steps: int = 50,
num_diffusion_samples: int = 1,
seed: int = 0,
) -> tuple[str, float, float, float]:
if sequence is None:
# default to the M.HhaI methyltransferase / DNA / SAH complex (PDB 1MHT);
# `C36` is the CCD code for 5-methylcytosine, `SAH` for the cofactor
spi = StructurePredictionInput(
sequences=[
ProteinInput(id="A", sequence=MHHAI_SEQUENCE),
DNAInput(
id="B",
sequence="GATAGCGCTATC",
modifications=[Modification(position=5, ccd="C36")],
),
DNAInput(
id="C",
sequence="TGATAGCGCTATC",
modifications=[Modification(position=6, ccd="C36")],
),
LigandInput(id="L", ccd=["SAH"]),
]
)
else:
spi = StructurePredictionInput(
sequences=[ProteinInput(id="A", sequence=sequence.strip())]
)
print(
f"🧬 folding with num_loops={num_loops}, "
f"num_sampling_steps={num_sampling_steps}, "
f"num_diffusion_samples={num_diffusion_samples}"
)
result = ESMFold2InputBuilder().fold(
self.model,
spi,
num_loops=num_loops,
num_sampling_steps=num_sampling_steps,
num_diffusion_samples=num_diffusion_samples,
seed=seed,
)
return (
result.complex.to_mmcif(),
float(result.plddt.mean()),
float(result.ptm),
float(result.iptm),
)Fold a complex from the command line
To showcase the full breadth of ESMFold2 — it can predict structures of proteins, nucleic acids, ligands, and modified residues all at once — we fold a complex by default: the M.HhaI cytosine-5 DNA methyltransferase from Haemophilus haemolyticus, bound to a methylated DNA duplex and the S-adenosyl-L-homocysteine cofactor that remains after methyl transfer.
MHHAI_SEQUENCE = (
"MIEIKDKQLTGLRFIDLFAGLGGFRLALESCGAECVYSNEWDKYAQEVYEMNFGEKPEGDITQVNEKTIPDH"
"DILCAGFPCQAFSISGKQKGFEDSRGTLFFDIARIVREKKPKVVFMENVKNFASHDNGNTLEVVKNTMNELD"
"YSFHAKVLNALDYGIPQKRERIYMICFRNDLNIQNFQFPKPFELNTFVKDLLLPDSEVEHLVIDRKDLVMTN"
"QEIEQTTPKTVRLGIVGKGGQGERIYSTRGIAITLSAYGGGIFAKTGGYLVNGKTRKLHPRECARVMGYPDS"
"YKVHPSTSQAYKQFGNSVVINVLQYIAYNIGSSLNFKPY"
)Fold the complex in the cloud by running the following command:
modal run esmfold2.pyThis will save the predicted structure locally as a Crystallographic Information File, which you can render with Mol* Viewer.
To fold a single protein chain instead, pass a sequence:
modal run esmfold2.py --sequence "MKTAYIAKQRQISFVKSHFSRQLEERLGLIEVQA..."@app.local_entrypoint()
def main(
sequence: Optional[str] = None,
output_path: Optional[str] = None,
):
print("🧬 running ESMFold2")
esmfold2 = ESMFold2Inference()
cif_text, plddt, ptm, iptm = esmfold2.fold.remote(sequence)
print(f"🧬 pLDDT mean: {plddt:.3f}, pTM: {ptm:.3f}, ipTM: {iptm:.3f}")
if output_path is None:
output_path = Path("/tmp") / "esmfold2" / "prediction.cif"
else:
output_path = Path(output_path)
output_path.parent.mkdir(parents=True, exist_ok=True)
print(f"🧬 writing predicted structure to {output_path}")
output_path.write_text(cif_text)